
diff options
Diffstat (limited to 'python-gmm-demux.spec')
| -rw-r--r-- | python-gmm-demux.spec | 866 |
1 files changed, 866 insertions, 0 deletions
diff --git a/python-gmm-demux.spec b/python-gmm-demux.spec new file mode 100644 index 0000000..045e04e --- /dev/null +++ b/python-gmm-demux.spec @@ -0,0 +1,866 @@ +%global _empty_manifest_terminate_build 0 +Name: python-GMM-Demux +Version: 0.2.1.3 +Release: 1 +Summary: A multiplet removal tool for processing cell hashing data +License: MIT License +URL: https://github.com/CHPGenetics/GMM-demux +Source0: https://mirrors.nju.edu.cn/pypi/web/packages/a4/e6/8cdb23eaf6e3c7b03a960acb11590ba6397deba9ac7258fc05ab2c7b855d/GMM_Demux-0.2.1.3.tar.gz +BuildArch: noarch + +Requires: python3-pandas +Requires: python3-numpy +Requires: python3-scipy +Requires: python3-tabulate +Requires: python3-argparse +Requires: python3-statistics +Requires: python3-BitVector +Requires: python3-sklearn + +%description +# GMM-Demux +GMM-Demux is a Gaussian-Mixture-Model-based software for processing sample barcoding data (cell hashing and MULTI-seq). + +GMM-Demux identifies Multi-Sample Multiplets (MSMs) in a sample barcoding dataset. Below shows an example distribution of MSMs in a PBMC scRNA-seq dataset. Orange dots in the scatter plot are MSMs. + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/GMM_simplified.png" alt="GMM-Demux example" width="600"/> + +## Description +GMM-Demux removes Multi-Sample-Multiplets (MSMs) in a cell hashing dataset and estimates the percentages of Same-Sample-Multiplets (SSMs) and singlets in the remaining dataset. +GMM-Demux also verifies if a putative cell type exists, or is it merely an artifact induced by multiplets. + +Multiplet-induced fake cell types are called "phony cell types". + +Examples of phony cell types in a PBMC CITE-seq dataset is provided in the figure below: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/phony.png" width="600"/> + +In the above figure, both the CD3+CD19+ and the CD4+CD8+ cell types are multiplet-induced fake cell types. + +Phony type clusters have large percentages of MSMs, as above figure shows. Both phony type clusters have large MSM percentages. + +Percentages of MSMs are used as key features by GMM-Demux to classify GEM clusters. + +## Terminology +* **Singlet**: A droplet that contains a single cell. + +* **MSM**: Multi-Sample Multiplet. A MSM is a multiplet that contains cells from different samples in sample barcoding. MSMs can be identified by GMM-Demux. + +* **SSM**: Same-Sample Multiplet. A SSM is a multiplet that contains cells from a single sample in sample barcoding. SSMs cannot be separated from singlets by sample barcoding. + +* **SSD**: Same-Sample Droplet. SSD is a combined category of both SSMs and singlets. + +* **Pure type**: a pure type cell type is a real cell type that exist in the tissue. + +* **Phony type**: a phony type cell type is an artificial cell type that is an artifact produced by multiplets. + +* **Mixture type**: a mixture type cell type is a cluster of droplets in which there exist a non-trivial fraction of phony type droplets. + +An illustration of the above terminologies in a PBMC dataset is provided in the figure below: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/term.png" width="600"/> + +## Features +* Remove cell-hashing-identifiable multiplets (i.e., MSMs) from the dataset. +* Estimate the fraction of cell-hashing-unidentifiable multiplets (SSMs) in the remaining dataset (the RSSM percentage). +* Test if a putative cell type is a pure (real) cell type or is it a phony (fake) cell type. + +## Example Dataset +* An example cell hashing dataset is provided in the *example_input* folder. It contains the per-drop HTO count matrix of a 4-sample cell hashing library prep. The input folder has the same file format with the CellRanger v3 output. + +# Authors + Hongyi Xin, Qi Yan, Yale Jiang, Jiadi Luo, Carla Erb, Richard Duerr, Kong Chen* and Wei Chen* + +# Maintainer +Hongyi Xin <gohongyi at gmail.edu> + +## Requirement + +GMM-Demux requires python3 (>3.5). + +## Install + +GMM-Demux can be directly installed from PyPi. Or it can be built and installed locally. + +### Install GMM-Demux from PyPi. +```bash +pip3 install --user GMM_Demux +``` + +In some OS, the `pip3` is linked to `pip` by default. For these OS, the installation command is simply: + +```bash +pip install --user GMM_Demux +``` + +Check if `pip3` is linked to `pip` with `pip -V`. + +If one chooses to install GMM-Demux from PyPi, it is unnecessary to download GMM-Demux from github. However, we still recommend downloading the example dataset to try out GMM-Demux. + +### Install GMM-Demux locally using [setuptools](https://packaging.python.org/tutorials/installing-packages/) and pip3. + +You may choose to install GMM-Demux locally after cloning the github repository. However, **this is for advanced users only and support is not gauranteed**. +The command is provided below: + +```bash +cd <GMM-Demux dir> +python3 setup.py sdist bdist_wheel +pip3 install --user . +``` + +### Post installation processes + +If this is the first time you install a python3 software through pip, make sure you add the pip binary folder to your `PATH` variable. +Typically, the pip binary folder is located at ```~/.local/bin```. + +The pip binary folder might locate at a different location if the user uses virtual enviroment. Pay attention to the pip installation output. + +Here is an example installation output. The path of the pip binary folder is highlighted: +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/path.png" width="500"/> + +To temporarily add the pip binary folder, run the following command: +```bash +export PATH=~/.local/bin:$PATH +``` + +To permenantly add the pip library folder to your `PATH` variable, append the following line to your `.bashrc` file (assuming bash is the default shell). +```bash +PATH=~/.local/bin:$PATH +``` + +## Content + +The source code of GMM-Demux is supplied in the ```GMM_Demux``` folder. + +An example cell hashing dataset is also provided, located in the ```example_input/outs/filtered_feature_bc_matrix``` folder. + +An example set of hand-curated putative cell types of the above dataset are provided in the ```example_cell_types``` folder. Cell types are annotated through manual gating using surface marker expression data. + +An example csv format of the above cell hashing dataset is provided as the ```example_hto.csv``` file. + +## Usage + +### Case 1: Basic Usage, Remove MSMs +Once installed, GMM-Demux is directly accessible with the ```GMM-demux``` command. +```bash +GMM-demux <cell_hashing_path> <HTO_names> +``` + +```<HTO_names>``` is a list of sample tags (HTOs) separated by ',' without whitespace. +For example, there are four sample barcoding tags in the example cell hashing dataset. +They are **HTO_1**, **HTO_2**, **HTO_3**, **HTO_4**. The ```<HTO_names>``` variable therefore is ```HTO_1,HTO_2,_HTO_3,HTO_4```. + +The non-MSM droplets (SSDs) of the dataset are stored in the *GMM_Demux_mtx* folder under the current directory by default. +The output path can also be specified through the `-o` flag. + +#### Example Command +An example cell hashing data is provided in the *example_input* folder. <HTO_names> can be obtained from the features.tsv file. +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 +``` + +<HTO_names> are included in the features.tsv file. The content of the feature.tsv file is shown below. + +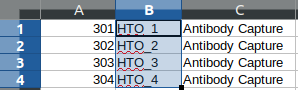 + +#### Output +The default content in the output folder are the non-MSM droplets (SSDs), stored in MTX format. The output shares the same format with CellRanger 3.0. By default, the output is stored in `SSD_mtx` folder. The output location can be overwritten with the `-o` flag. + +### Case 2: Compute the MSM and SSM rates +To compute the MSM and SSM rates, GMM-Demux requires the `-u` flag: + +* -u SUMMARY, --summary SUMMARY Generate the statstic summary of the dataset. Requires an estimated total number of cells in the assay as input. + +The `-u` flag requires an additional <NUM_OF_CELL> argument, which is the estimated total count of cells in the single cell assay. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 +``` + +#### Output +Below is an example report: + + +* RSSM denotes the percentage of SSMs among the remaining SSDs (after removing all MSMs). RSSM **measures the quality of the final cell hashing dataset after removing MSMs**. + +### Case 3: Verify if a cell type exists +GMM-Demux verifies if a putative cell type exists with the `-e` flag: + +* -e EXAMINE, --examine EXAMINE Provide the cell list. Requires a file argument. Only executes if -u is set. + +The `-e` flag requires a file name, which stores the list of droplet barcodes of the putative cell type. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 -e example_cell_types/CD19+.txt +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 -e example_cell_types/Doublets/CD3+CD4+CD19+.txt +``` + +#### Output +An example output of a pure cell type: +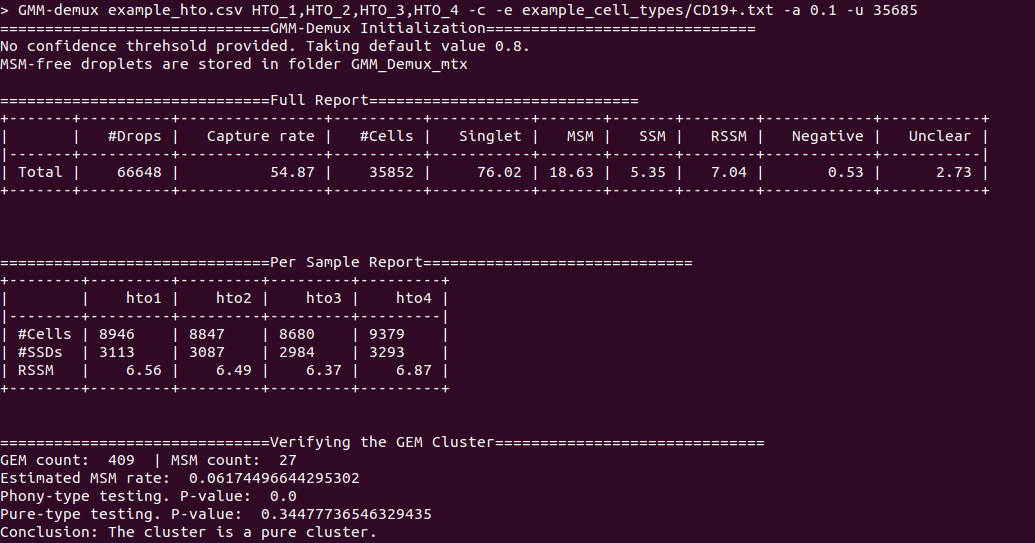 + +An example output of a phony cell type: +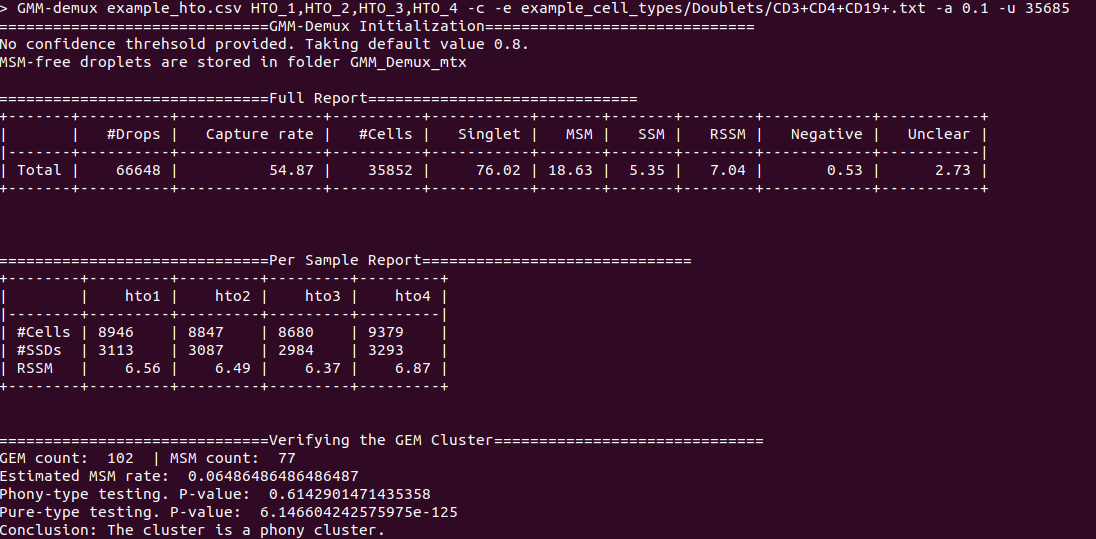 + +### Case 4: Use the csv file format as input, instead of the mtx format +#### Example Command +```bash +GMM-demux -c example_hto.csv HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 +``` + +### Case 5: Extract droplets that are labeled by a combination of sample tags +Extract droplets that are labeled by multiple sample barcoding tags, with the `-x` flag: + +* -x EXTRACT, --extract EXTRACT Names of the sample barcoding tag(s) to extract, separated by ','. Joint tags are linked with '+'. + +**When `-x` is set, other functions of GMM-Demux will be turned off.** + +#### *Case 5a: Extract a single HTO sample* + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO_1 +``` +#### *Case 5b: Extract a single HTO sample that are jointly defined by multiple HTO tags* +Use `+` to specify the joint HTO tags. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO_1+HTO_2 +``` +#### *Case 5c: Extract multiple HTO samples* +Use `,` to separate sample tags. Single tag samples can be merged with joint-tag samples. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO3,HTO_1+HTO_2,HTO_1+HTO_4+HTO_2 +``` + +## Optional Arguments +* -h: show help information. +* -f FULL, --full FULL Generate the full classification report. Require a path argument. +* -s SIMPLIFIED, --simplified SIMPLIFIED Generate the simplified classification report. Require a path argument. +* -o OUTPUT, --output OUTPUT The path for storing the Same-Sample-Droplets (SSDs). SSDs are stored in mtx format. Requires a path argument. Default path: SSD_mtx. +* -r REPORT, --report REPORT Specify the file to store summary report. Require a file argument. +* -c CSV, --csv Take input in csv format, instead of mmx format. +* -s SKIP, --skip FULL\_REPORT Load a full classification report and skip the mtx folder as input. Require a path argument. +* -a AMBIGUOUS, --ambiguous AMBIGUOUS The estimated chance of having a phony GEM getting included in a pure type GEM cluster by the clustering algorithm. Requires a float in (0, 1). Default value: 0.05. Only executes if -e executes. +* -t THRESHOLD, --threshold THRESHOLD Provide the confidence threshold value. Requires a float in (0,1). Default value: 0.8. + +## Parsing the Classification Output +There are two files in a classification output folder. A config file (ending with .config) and a classification file (ending with .csv). + +The classification file contains the label of each droplet as well as the probability of the classification. The classification is represented with numbers which are explained in the config file. + +Below shows the classification output of the example data: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/class_output.png" width="600"/> + +## Online Cell Hashing Experiment Planner +A GMM-Demux based online cell hashing experiment planner is publically accessible at [here](https://www.pitt.edu/~wec47/gmmdemux.html). + +[<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/planner.png" alt="Online explanner example" width="600"/>](https://www.pitt.edu/~wec47/gmmdemux.html) + +## Citation +If you find this code useful in your research, please consider citing: + + @article{xin2019sample, + title={Sample demultiplexing, multiplet detection, experiment planning and novel cell type verification in single cell sequencing}, + author={Xin, Hongyi and Yan, Qi and Jiang, Yale and Lian, Qiuyu and Luo, Jiadi and Erb, Carla and Duerr, Richard and Chen, Kong and Chen, Wei}, + journal={bioRxiv}, + pages={828483}, + year={2019}, + publisher={Cold Spring Harbor Laboratory} + } + +## Acknowledgement + +Special thank to Zhongli Xu for testing GMM-Demux! + + + + +%package -n python3-GMM-Demux +Summary: A multiplet removal tool for processing cell hashing data +Provides: python-GMM-Demux +BuildRequires: python3-devel +BuildRequires: python3-setuptools +BuildRequires: python3-pip +%description -n python3-GMM-Demux +# GMM-Demux +GMM-Demux is a Gaussian-Mixture-Model-based software for processing sample barcoding data (cell hashing and MULTI-seq). + +GMM-Demux identifies Multi-Sample Multiplets (MSMs) in a sample barcoding dataset. Below shows an example distribution of MSMs in a PBMC scRNA-seq dataset. Orange dots in the scatter plot are MSMs. + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/GMM_simplified.png" alt="GMM-Demux example" width="600"/> + +## Description +GMM-Demux removes Multi-Sample-Multiplets (MSMs) in a cell hashing dataset and estimates the percentages of Same-Sample-Multiplets (SSMs) and singlets in the remaining dataset. +GMM-Demux also verifies if a putative cell type exists, or is it merely an artifact induced by multiplets. + +Multiplet-induced fake cell types are called "phony cell types". + +Examples of phony cell types in a PBMC CITE-seq dataset is provided in the figure below: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/phony.png" width="600"/> + +In the above figure, both the CD3+CD19+ and the CD4+CD8+ cell types are multiplet-induced fake cell types. + +Phony type clusters have large percentages of MSMs, as above figure shows. Both phony type clusters have large MSM percentages. + +Percentages of MSMs are used as key features by GMM-Demux to classify GEM clusters. + +## Terminology +* **Singlet**: A droplet that contains a single cell. + +* **MSM**: Multi-Sample Multiplet. A MSM is a multiplet that contains cells from different samples in sample barcoding. MSMs can be identified by GMM-Demux. + +* **SSM**: Same-Sample Multiplet. A SSM is a multiplet that contains cells from a single sample in sample barcoding. SSMs cannot be separated from singlets by sample barcoding. + +* **SSD**: Same-Sample Droplet. SSD is a combined category of both SSMs and singlets. + +* **Pure type**: a pure type cell type is a real cell type that exist in the tissue. + +* **Phony type**: a phony type cell type is an artificial cell type that is an artifact produced by multiplets. + +* **Mixture type**: a mixture type cell type is a cluster of droplets in which there exist a non-trivial fraction of phony type droplets. + +An illustration of the above terminologies in a PBMC dataset is provided in the figure below: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/term.png" width="600"/> + +## Features +* Remove cell-hashing-identifiable multiplets (i.e., MSMs) from the dataset. +* Estimate the fraction of cell-hashing-unidentifiable multiplets (SSMs) in the remaining dataset (the RSSM percentage). +* Test if a putative cell type is a pure (real) cell type or is it a phony (fake) cell type. + +## Example Dataset +* An example cell hashing dataset is provided in the *example_input* folder. It contains the per-drop HTO count matrix of a 4-sample cell hashing library prep. The input folder has the same file format with the CellRanger v3 output. + +# Authors + Hongyi Xin, Qi Yan, Yale Jiang, Jiadi Luo, Carla Erb, Richard Duerr, Kong Chen* and Wei Chen* + +# Maintainer +Hongyi Xin <gohongyi at gmail.edu> + +## Requirement + +GMM-Demux requires python3 (>3.5). + +## Install + +GMM-Demux can be directly installed from PyPi. Or it can be built and installed locally. + +### Install GMM-Demux from PyPi. +```bash +pip3 install --user GMM_Demux +``` + +In some OS, the `pip3` is linked to `pip` by default. For these OS, the installation command is simply: + +```bash +pip install --user GMM_Demux +``` + +Check if `pip3` is linked to `pip` with `pip -V`. + +If one chooses to install GMM-Demux from PyPi, it is unnecessary to download GMM-Demux from github. However, we still recommend downloading the example dataset to try out GMM-Demux. + +### Install GMM-Demux locally using [setuptools](https://packaging.python.org/tutorials/installing-packages/) and pip3. + +You may choose to install GMM-Demux locally after cloning the github repository. However, **this is for advanced users only and support is not gauranteed**. +The command is provided below: + +```bash +cd <GMM-Demux dir> +python3 setup.py sdist bdist_wheel +pip3 install --user . +``` + +### Post installation processes + +If this is the first time you install a python3 software through pip, make sure you add the pip binary folder to your `PATH` variable. +Typically, the pip binary folder is located at ```~/.local/bin```. + +The pip binary folder might locate at a different location if the user uses virtual enviroment. Pay attention to the pip installation output. + +Here is an example installation output. The path of the pip binary folder is highlighted: +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/path.png" width="500"/> + +To temporarily add the pip binary folder, run the following command: +```bash +export PATH=~/.local/bin:$PATH +``` + +To permenantly add the pip library folder to your `PATH` variable, append the following line to your `.bashrc` file (assuming bash is the default shell). +```bash +PATH=~/.local/bin:$PATH +``` + +## Content + +The source code of GMM-Demux is supplied in the ```GMM_Demux``` folder. + +An example cell hashing dataset is also provided, located in the ```example_input/outs/filtered_feature_bc_matrix``` folder. + +An example set of hand-curated putative cell types of the above dataset are provided in the ```example_cell_types``` folder. Cell types are annotated through manual gating using surface marker expression data. + +An example csv format of the above cell hashing dataset is provided as the ```example_hto.csv``` file. + +## Usage + +### Case 1: Basic Usage, Remove MSMs +Once installed, GMM-Demux is directly accessible with the ```GMM-demux``` command. +```bash +GMM-demux <cell_hashing_path> <HTO_names> +``` + +```<HTO_names>``` is a list of sample tags (HTOs) separated by ',' without whitespace. +For example, there are four sample barcoding tags in the example cell hashing dataset. +They are **HTO_1**, **HTO_2**, **HTO_3**, **HTO_4**. The ```<HTO_names>``` variable therefore is ```HTO_1,HTO_2,_HTO_3,HTO_4```. + +The non-MSM droplets (SSDs) of the dataset are stored in the *GMM_Demux_mtx* folder under the current directory by default. +The output path can also be specified through the `-o` flag. + +#### Example Command +An example cell hashing data is provided in the *example_input* folder. <HTO_names> can be obtained from the features.tsv file. +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 +``` + +<HTO_names> are included in the features.tsv file. The content of the feature.tsv file is shown below. + + + +#### Output +The default content in the output folder are the non-MSM droplets (SSDs), stored in MTX format. The output shares the same format with CellRanger 3.0. By default, the output is stored in `SSD_mtx` folder. The output location can be overwritten with the `-o` flag. + +### Case 2: Compute the MSM and SSM rates +To compute the MSM and SSM rates, GMM-Demux requires the `-u` flag: + +* -u SUMMARY, --summary SUMMARY Generate the statstic summary of the dataset. Requires an estimated total number of cells in the assay as input. + +The `-u` flag requires an additional <NUM_OF_CELL> argument, which is the estimated total count of cells in the single cell assay. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 +``` + +#### Output +Below is an example report: + + +* RSSM denotes the percentage of SSMs among the remaining SSDs (after removing all MSMs). RSSM **measures the quality of the final cell hashing dataset after removing MSMs**. + +### Case 3: Verify if a cell type exists +GMM-Demux verifies if a putative cell type exists with the `-e` flag: + +* -e EXAMINE, --examine EXAMINE Provide the cell list. Requires a file argument. Only executes if -u is set. + +The `-e` flag requires a file name, which stores the list of droplet barcodes of the putative cell type. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 -e example_cell_types/CD19+.txt +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 -e example_cell_types/Doublets/CD3+CD4+CD19+.txt +``` + +#### Output +An example output of a pure cell type: + + +An example output of a phony cell type: + + +### Case 4: Use the csv file format as input, instead of the mtx format +#### Example Command +```bash +GMM-demux -c example_hto.csv HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 +``` + +### Case 5: Extract droplets that are labeled by a combination of sample tags +Extract droplets that are labeled by multiple sample barcoding tags, with the `-x` flag: + +* -x EXTRACT, --extract EXTRACT Names of the sample barcoding tag(s) to extract, separated by ','. Joint tags are linked with '+'. + +**When `-x` is set, other functions of GMM-Demux will be turned off.** + +#### *Case 5a: Extract a single HTO sample* + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO_1 +``` +#### *Case 5b: Extract a single HTO sample that are jointly defined by multiple HTO tags* +Use `+` to specify the joint HTO tags. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO_1+HTO_2 +``` +#### *Case 5c: Extract multiple HTO samples* +Use `,` to separate sample tags. Single tag samples can be merged with joint-tag samples. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO3,HTO_1+HTO_2,HTO_1+HTO_4+HTO_2 +``` + +## Optional Arguments +* -h: show help information. +* -f FULL, --full FULL Generate the full classification report. Require a path argument. +* -s SIMPLIFIED, --simplified SIMPLIFIED Generate the simplified classification report. Require a path argument. +* -o OUTPUT, --output OUTPUT The path for storing the Same-Sample-Droplets (SSDs). SSDs are stored in mtx format. Requires a path argument. Default path: SSD_mtx. +* -r REPORT, --report REPORT Specify the file to store summary report. Require a file argument. +* -c CSV, --csv Take input in csv format, instead of mmx format. +* -s SKIP, --skip FULL\_REPORT Load a full classification report and skip the mtx folder as input. Require a path argument. +* -a AMBIGUOUS, --ambiguous AMBIGUOUS The estimated chance of having a phony GEM getting included in a pure type GEM cluster by the clustering algorithm. Requires a float in (0, 1). Default value: 0.05. Only executes if -e executes. +* -t THRESHOLD, --threshold THRESHOLD Provide the confidence threshold value. Requires a float in (0,1). Default value: 0.8. + +## Parsing the Classification Output +There are two files in a classification output folder. A config file (ending with .config) and a classification file (ending with .csv). + +The classification file contains the label of each droplet as well as the probability of the classification. The classification is represented with numbers which are explained in the config file. + +Below shows the classification output of the example data: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/class_output.png" width="600"/> + +## Online Cell Hashing Experiment Planner +A GMM-Demux based online cell hashing experiment planner is publically accessible at [here](https://www.pitt.edu/~wec47/gmmdemux.html). + +[<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/planner.png" alt="Online explanner example" width="600"/>](https://www.pitt.edu/~wec47/gmmdemux.html) + +## Citation +If you find this code useful in your research, please consider citing: + + @article{xin2019sample, + title={Sample demultiplexing, multiplet detection, experiment planning and novel cell type verification in single cell sequencing}, + author={Xin, Hongyi and Yan, Qi and Jiang, Yale and Lian, Qiuyu and Luo, Jiadi and Erb, Carla and Duerr, Richard and Chen, Kong and Chen, Wei}, + journal={bioRxiv}, + pages={828483}, + year={2019}, + publisher={Cold Spring Harbor Laboratory} + } + +## Acknowledgement + +Special thank to Zhongli Xu for testing GMM-Demux! + + + + +%package help +Summary: Development documents and examples for GMM-Demux +Provides: python3-GMM-Demux-doc +%description help +# GMM-Demux +GMM-Demux is a Gaussian-Mixture-Model-based software for processing sample barcoding data (cell hashing and MULTI-seq). + +GMM-Demux identifies Multi-Sample Multiplets (MSMs) in a sample barcoding dataset. Below shows an example distribution of MSMs in a PBMC scRNA-seq dataset. Orange dots in the scatter plot are MSMs. + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/GMM_simplified.png" alt="GMM-Demux example" width="600"/> + +## Description +GMM-Demux removes Multi-Sample-Multiplets (MSMs) in a cell hashing dataset and estimates the percentages of Same-Sample-Multiplets (SSMs) and singlets in the remaining dataset. +GMM-Demux also verifies if a putative cell type exists, or is it merely an artifact induced by multiplets. + +Multiplet-induced fake cell types are called "phony cell types". + +Examples of phony cell types in a PBMC CITE-seq dataset is provided in the figure below: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/phony.png" width="600"/> + +In the above figure, both the CD3+CD19+ and the CD4+CD8+ cell types are multiplet-induced fake cell types. + +Phony type clusters have large percentages of MSMs, as above figure shows. Both phony type clusters have large MSM percentages. + +Percentages of MSMs are used as key features by GMM-Demux to classify GEM clusters. + +## Terminology +* **Singlet**: A droplet that contains a single cell. + +* **MSM**: Multi-Sample Multiplet. A MSM is a multiplet that contains cells from different samples in sample barcoding. MSMs can be identified by GMM-Demux. + +* **SSM**: Same-Sample Multiplet. A SSM is a multiplet that contains cells from a single sample in sample barcoding. SSMs cannot be separated from singlets by sample barcoding. + +* **SSD**: Same-Sample Droplet. SSD is a combined category of both SSMs and singlets. + +* **Pure type**: a pure type cell type is a real cell type that exist in the tissue. + +* **Phony type**: a phony type cell type is an artificial cell type that is an artifact produced by multiplets. + +* **Mixture type**: a mixture type cell type is a cluster of droplets in which there exist a non-trivial fraction of phony type droplets. + +An illustration of the above terminologies in a PBMC dataset is provided in the figure below: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/term.png" width="600"/> + +## Features +* Remove cell-hashing-identifiable multiplets (i.e., MSMs) from the dataset. +* Estimate the fraction of cell-hashing-unidentifiable multiplets (SSMs) in the remaining dataset (the RSSM percentage). +* Test if a putative cell type is a pure (real) cell type or is it a phony (fake) cell type. + +## Example Dataset +* An example cell hashing dataset is provided in the *example_input* folder. It contains the per-drop HTO count matrix of a 4-sample cell hashing library prep. The input folder has the same file format with the CellRanger v3 output. + +# Authors + Hongyi Xin, Qi Yan, Yale Jiang, Jiadi Luo, Carla Erb, Richard Duerr, Kong Chen* and Wei Chen* + +# Maintainer +Hongyi Xin <gohongyi at gmail.edu> + +## Requirement + +GMM-Demux requires python3 (>3.5). + +## Install + +GMM-Demux can be directly installed from PyPi. Or it can be built and installed locally. + +### Install GMM-Demux from PyPi. +```bash +pip3 install --user GMM_Demux +``` + +In some OS, the `pip3` is linked to `pip` by default. For these OS, the installation command is simply: + +```bash +pip install --user GMM_Demux +``` + +Check if `pip3` is linked to `pip` with `pip -V`. + +If one chooses to install GMM-Demux from PyPi, it is unnecessary to download GMM-Demux from github. However, we still recommend downloading the example dataset to try out GMM-Demux. + +### Install GMM-Demux locally using [setuptools](https://packaging.python.org/tutorials/installing-packages/) and pip3. + +You may choose to install GMM-Demux locally after cloning the github repository. However, **this is for advanced users only and support is not gauranteed**. +The command is provided below: + +```bash +cd <GMM-Demux dir> +python3 setup.py sdist bdist_wheel +pip3 install --user . +``` + +### Post installation processes + +If this is the first time you install a python3 software through pip, make sure you add the pip binary folder to your `PATH` variable. +Typically, the pip binary folder is located at ```~/.local/bin```. + +The pip binary folder might locate at a different location if the user uses virtual enviroment. Pay attention to the pip installation output. + +Here is an example installation output. The path of the pip binary folder is highlighted: +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/path.png" width="500"/> + +To temporarily add the pip binary folder, run the following command: +```bash +export PATH=~/.local/bin:$PATH +``` + +To permenantly add the pip library folder to your `PATH` variable, append the following line to your `.bashrc` file (assuming bash is the default shell). +```bash +PATH=~/.local/bin:$PATH +``` + +## Content + +The source code of GMM-Demux is supplied in the ```GMM_Demux``` folder. + +An example cell hashing dataset is also provided, located in the ```example_input/outs/filtered_feature_bc_matrix``` folder. + +An example set of hand-curated putative cell types of the above dataset are provided in the ```example_cell_types``` folder. Cell types are annotated through manual gating using surface marker expression data. + +An example csv format of the above cell hashing dataset is provided as the ```example_hto.csv``` file. + +## Usage + +### Case 1: Basic Usage, Remove MSMs +Once installed, GMM-Demux is directly accessible with the ```GMM-demux``` command. +```bash +GMM-demux <cell_hashing_path> <HTO_names> +``` + +```<HTO_names>``` is a list of sample tags (HTOs) separated by ',' without whitespace. +For example, there are four sample barcoding tags in the example cell hashing dataset. +They are **HTO_1**, **HTO_2**, **HTO_3**, **HTO_4**. The ```<HTO_names>``` variable therefore is ```HTO_1,HTO_2,_HTO_3,HTO_4```. + +The non-MSM droplets (SSDs) of the dataset are stored in the *GMM_Demux_mtx* folder under the current directory by default. +The output path can also be specified through the `-o` flag. + +#### Example Command +An example cell hashing data is provided in the *example_input* folder. <HTO_names> can be obtained from the features.tsv file. +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 +``` + +<HTO_names> are included in the features.tsv file. The content of the feature.tsv file is shown below. + + + +#### Output +The default content in the output folder are the non-MSM droplets (SSDs), stored in MTX format. The output shares the same format with CellRanger 3.0. By default, the output is stored in `SSD_mtx` folder. The output location can be overwritten with the `-o` flag. + +### Case 2: Compute the MSM and SSM rates +To compute the MSM and SSM rates, GMM-Demux requires the `-u` flag: + +* -u SUMMARY, --summary SUMMARY Generate the statstic summary of the dataset. Requires an estimated total number of cells in the assay as input. + +The `-u` flag requires an additional <NUM_OF_CELL> argument, which is the estimated total count of cells in the single cell assay. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 +``` + +#### Output +Below is an example report: + + +* RSSM denotes the percentage of SSMs among the remaining SSDs (after removing all MSMs). RSSM **measures the quality of the final cell hashing dataset after removing MSMs**. + +### Case 3: Verify if a cell type exists +GMM-Demux verifies if a putative cell type exists with the `-e` flag: + +* -e EXAMINE, --examine EXAMINE Provide the cell list. Requires a file argument. Only executes if -u is set. + +The `-e` flag requires a file name, which stores the list of droplet barcodes of the putative cell type. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 -e example_cell_types/CD19+.txt +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 -e example_cell_types/Doublets/CD3+CD4+CD19+.txt +``` + +#### Output +An example output of a pure cell type: + + +An example output of a phony cell type: + + +### Case 4: Use the csv file format as input, instead of the mtx format +#### Example Command +```bash +GMM-demux -c example_hto.csv HTO_1,HTO_2,HTO_3,HTO_4 -u 35685 +``` + +### Case 5: Extract droplets that are labeled by a combination of sample tags +Extract droplets that are labeled by multiple sample barcoding tags, with the `-x` flag: + +* -x EXTRACT, --extract EXTRACT Names of the sample barcoding tag(s) to extract, separated by ','. Joint tags are linked with '+'. + +**When `-x` is set, other functions of GMM-Demux will be turned off.** + +#### *Case 5a: Extract a single HTO sample* + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO_1 +``` +#### *Case 5b: Extract a single HTO sample that are jointly defined by multiple HTO tags* +Use `+` to specify the joint HTO tags. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO_1+HTO_2 +``` +#### *Case 5c: Extract multiple HTO samples* +Use `,` to separate sample tags. Single tag samples can be merged with joint-tag samples. + +#### Example Command +```bash +GMM-demux example_input/outs/filtered_feature_bc_matrix HTO_1,HTO_2,HTO_3,HTO_4 -x HTO3,HTO_1+HTO_2,HTO_1+HTO_4+HTO_2 +``` + +## Optional Arguments +* -h: show help information. +* -f FULL, --full FULL Generate the full classification report. Require a path argument. +* -s SIMPLIFIED, --simplified SIMPLIFIED Generate the simplified classification report. Require a path argument. +* -o OUTPUT, --output OUTPUT The path for storing the Same-Sample-Droplets (SSDs). SSDs are stored in mtx format. Requires a path argument. Default path: SSD_mtx. +* -r REPORT, --report REPORT Specify the file to store summary report. Require a file argument. +* -c CSV, --csv Take input in csv format, instead of mmx format. +* -s SKIP, --skip FULL\_REPORT Load a full classification report and skip the mtx folder as input. Require a path argument. +* -a AMBIGUOUS, --ambiguous AMBIGUOUS The estimated chance of having a phony GEM getting included in a pure type GEM cluster by the clustering algorithm. Requires a float in (0, 1). Default value: 0.05. Only executes if -e executes. +* -t THRESHOLD, --threshold THRESHOLD Provide the confidence threshold value. Requires a float in (0,1). Default value: 0.8. + +## Parsing the Classification Output +There are two files in a classification output folder. A config file (ending with .config) and a classification file (ending with .csv). + +The classification file contains the label of each droplet as well as the probability of the classification. The classification is represented with numbers which are explained in the config file. + +Below shows the classification output of the example data: + +<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/class_output.png" width="600"/> + +## Online Cell Hashing Experiment Planner +A GMM-Demux based online cell hashing experiment planner is publically accessible at [here](https://www.pitt.edu/~wec47/gmmdemux.html). + +[<img src="https://raw.githubusercontent.com/CHPGenetics/GMM-Demux/master/planner.png" alt="Online explanner example" width="600"/>](https://www.pitt.edu/~wec47/gmmdemux.html) + +## Citation +If you find this code useful in your research, please consider citing: + + @article{xin2019sample, + title={Sample demultiplexing, multiplet detection, experiment planning and novel cell type verification in single cell sequencing}, + author={Xin, Hongyi and Yan, Qi and Jiang, Yale and Lian, Qiuyu and Luo, Jiadi and Erb, Carla and Duerr, Richard and Chen, Kong and Chen, Wei}, + journal={bioRxiv}, + pages={828483}, + year={2019}, + publisher={Cold Spring Harbor Laboratory} + } + +## Acknowledgement + +Special thank to Zhongli Xu for testing GMM-Demux! + + + + +%prep +%autosetup -n GMM-Demux-0.2.1.3 + +%build +%py3_build + +%install +%py3_install +install -d -m755 %{buildroot}/%{_pkgdocdir} +if [ -d doc ]; then cp -arf doc %{buildroot}/%{_pkgdocdir}; fi +if [ -d docs ]; then cp -arf docs %{buildroot}/%{_pkgdocdir}; fi +if [ -d example ]; then cp -arf example %{buildroot}/%{_pkgdocdir}; fi +if [ -d examples ]; then cp -arf examples %{buildroot}/%{_pkgdocdir}; fi +pushd %{buildroot} +if [ -d usr/lib ]; then + find usr/lib -type f -printf "/%h/%f\n" >> filelist.lst +fi +if [ -d usr/lib64 ]; then + find usr/lib64 -type f -printf "/%h/%f\n" >> filelist.lst +fi +if [ -d usr/bin ]; then + find usr/bin -type f -printf "/%h/%f\n" >> filelist.lst +fi +if [ -d usr/sbin ]; then + find usr/sbin -type f -printf "/%h/%f\n" >> filelist.lst +fi +touch doclist.lst +if [ -d usr/share/man ]; then + find usr/share/man -type f -printf "/%h/%f.gz\n" >> doclist.lst +fi +popd +mv %{buildroot}/filelist.lst . +mv %{buildroot}/doclist.lst . + +%files -n python3-GMM-Demux -f filelist.lst +%dir %{python3_sitelib}/* + +%files help -f doclist.lst +%{_docdir}/* + +%changelog +* Mon May 29 2023 Python_Bot <Python_Bot@openeuler.org> - 0.2.1.3-1 +- Package Spec generated |